Allosteric Drug Design in Proteases
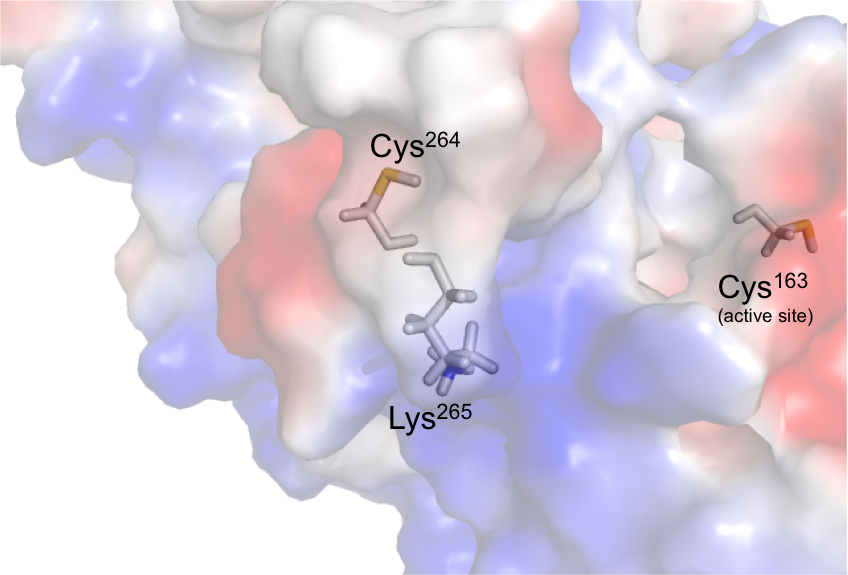
Caspases, a family of cysteine proteases, play essential roles in apoptosis, Inflammation and neurodegeneration. Inhibiting only one caspase over the other caspases is a challenging using active site (orthosteric) inhibitors because some of the caspases are closely related to each other. For example caspase-6 is closely related structurally to caspase-3 and -7, but plays very unique roles in human disease. Caspase-6, a protein that we have determined the structure of by x-ray crystallography, cleaves many proteins involved in neurodegeneration: tau, amyloid precursor protein (APP), Huntingtin protein (HTT). To date the most selective reported casp-6 inhibitors are only 10-fold selective over the most closely related caspases. We have discovered unique casp-6 inhibitors that are 500 to 10,000-fold selective over the two most closely related caspases. These inhibitors are extremely selective because they bind allosterically to cys264, a residue which is present only on Loop-4 in casp-6 but not in any other caspase. We are working to apply these inhibitors to the treatment of human diseases including Alzheimer’s Disease and Huntington’s Disease.
Flaviviruses – Structure and Drug Design
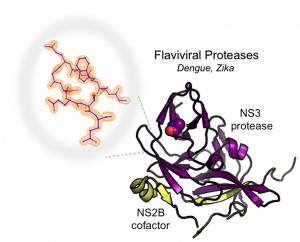
Flaviviruses, including dengue virus and Zika virus, contain a positive-sense RNA genome that is expressed as a single polyprotein precursor. Production of the active virus is dependent on the viral protease (NS2B-NS3pro) to cleave the polyprotein. As such, the protease is an excellent target to treat individuals who have been infected. Despite this knowledge, no protease-directed therapies have yet emerged, likely because most drug discovery efforts have focused on the active site. We are interested in studying allosteric sites on these viral proteases as an alternative way to inhibit their function. We have already identified a novel allosteric site centered at A125 that blocks the motion of a critical loop required for proteolytic activity. Our current research efforts are focused on discovering allosteric compounds that lock the enzyme into an inactive conformation and crystallizing such states to better understand the mechanism of inhibition. We are actively pursuing development of inhibitors for a number of viral proteases for therapeutic use.
Measuring Protease Dynamics Using H/DX-MS
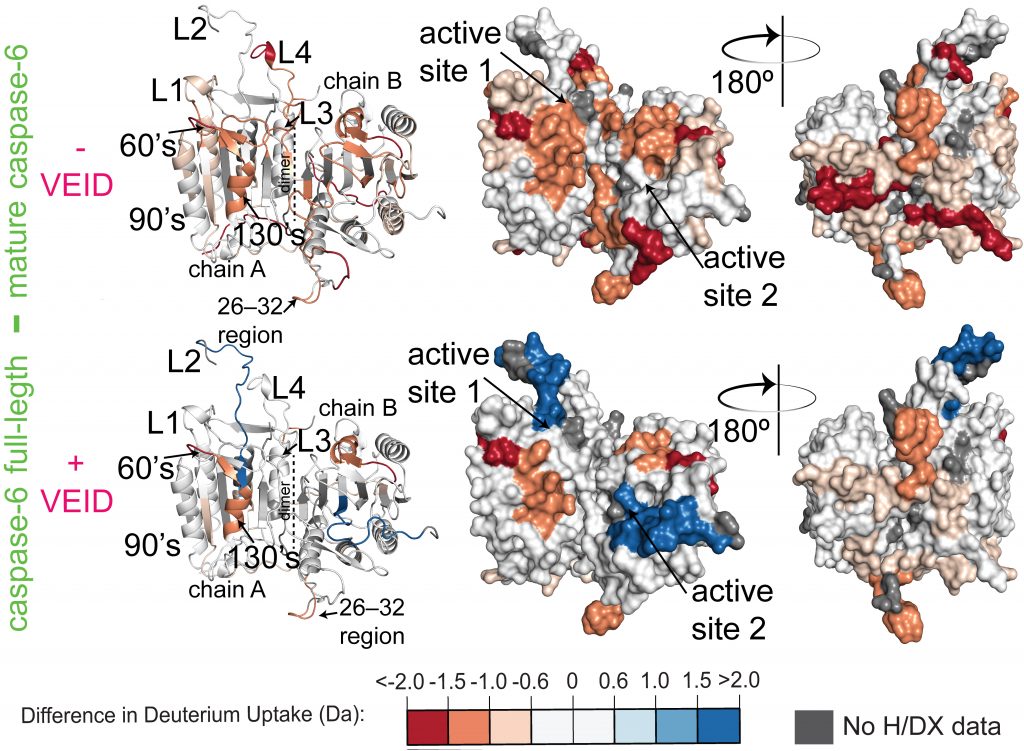
Caspase-6 is a dimeric cysteine aspartate protease involved in multiple biological pathways including apoptosis and neurodegeration. It is composed of prodomain (resi. 1-23), large subunit (resi. 24-179), intersubunit linker (resi. 179-193), and small subunit (resi. 193-293) for each monomer. Removal of the prodomain as well as intersubunit linker is a perquisite for caspase-6 maturation and then substrate binding. Multiple crystal structures of caspase-6 have been solved by the Hardy group and others.[1-3] However, these crystal structures lack structural-dynamic information. The Hardy Group has employed H/DX-MS technique to identify most important regions of caspase-6 upon substrate binding (here, VEID-cho). The H/DX data demonstrates that the active sites along with the dimer interface (consisting intersubunit linker) are highly exposed (colored red) to the solvent in the absence of VEID-cho. Moreover, drastic dynamic changes have been observed for these regions as unexposed (shifting from red to blue) to the solvent in the presence of VEID-cho. This structural-dynamic study will be helpful in identifying allosteric sites and therefore in designing potent as well as selective inhibitors of caspase-6.
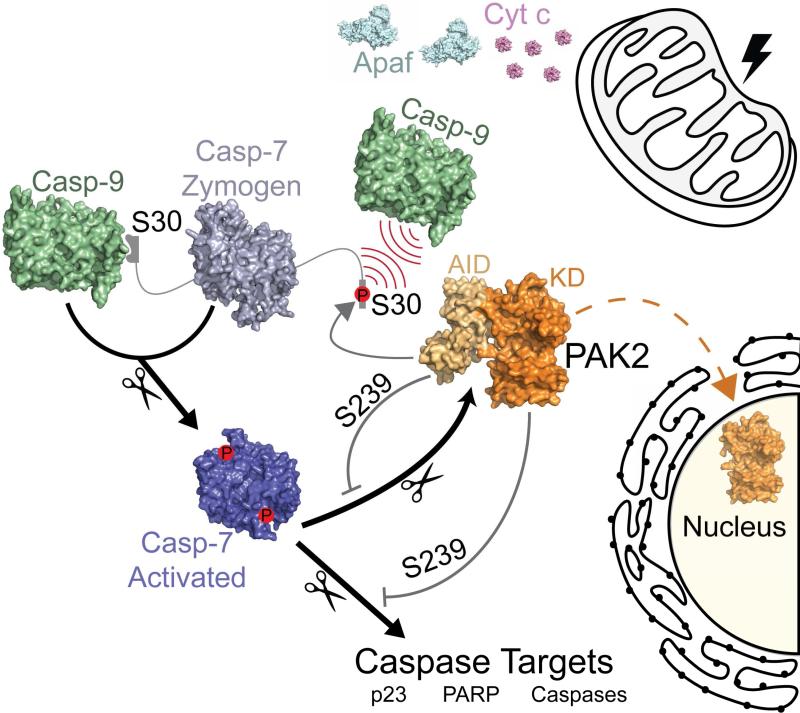
Determining Caspase Functionality Upon Phosphorylation
The Hardy Lab is very interested in identifying and exploiting allosteric sites. One of the richest sources of information about allosteric sites that we have developed thus far is probing the historical record of functional sites of phosphorylation. By studying how phosphorylation impacts caspase function, we have uncovered four new mechanisms by which phosphorylationimpacts caspase function. Phosphorylation disrupts loop conformation and prevents substrate binding (caspase-6), prevents recognition by an upstream caspase (caspase-7), directly blocks substrate binding (caspase-9) and leads to unfolding of the caspase-9 core in another case. These studies not only improve our understanding of caspase biology, they also provide ideas for exploitable mechanisms for controlling caspase function.
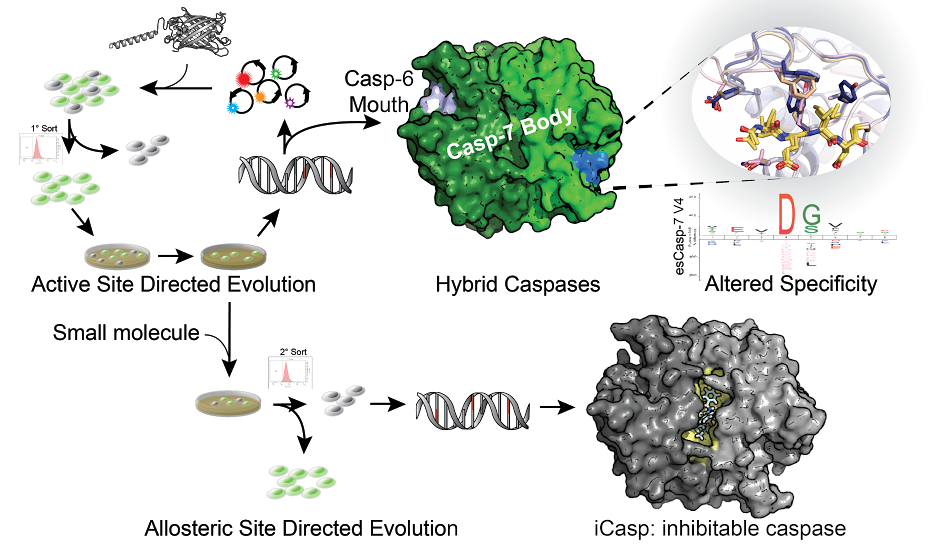
Engineering Active and Allosteric Sites
Our lab has developed a directed evolution method using our dark-to-bright GFP reporter that enables us to to tune specificity into intracellular proteases. In our first application of this technology, we sought to change the specificity of caspase-7 (DEVD) to that of caspase-6 (VEID) and produced evolved-specificity caspase-7 (esCasp-7) variants that maintained the body of caspase-7, but had caspase-6-like activity. With this hybrid enzyme, we sought to decipher proteins that may rely on non-active site interactions (exosites) for recognition. Such reprogrammed caspases represent a new method to discover exosite driven substrates and should be broadly applicable to many other proteases. We are also interested in engineering allosteric sites on caspases to develop iCasps: caspases that are inhibited by a small molecule of interest. This type of regulation is desirable so that caspase activity can be silenced only under specific cellular conditions.
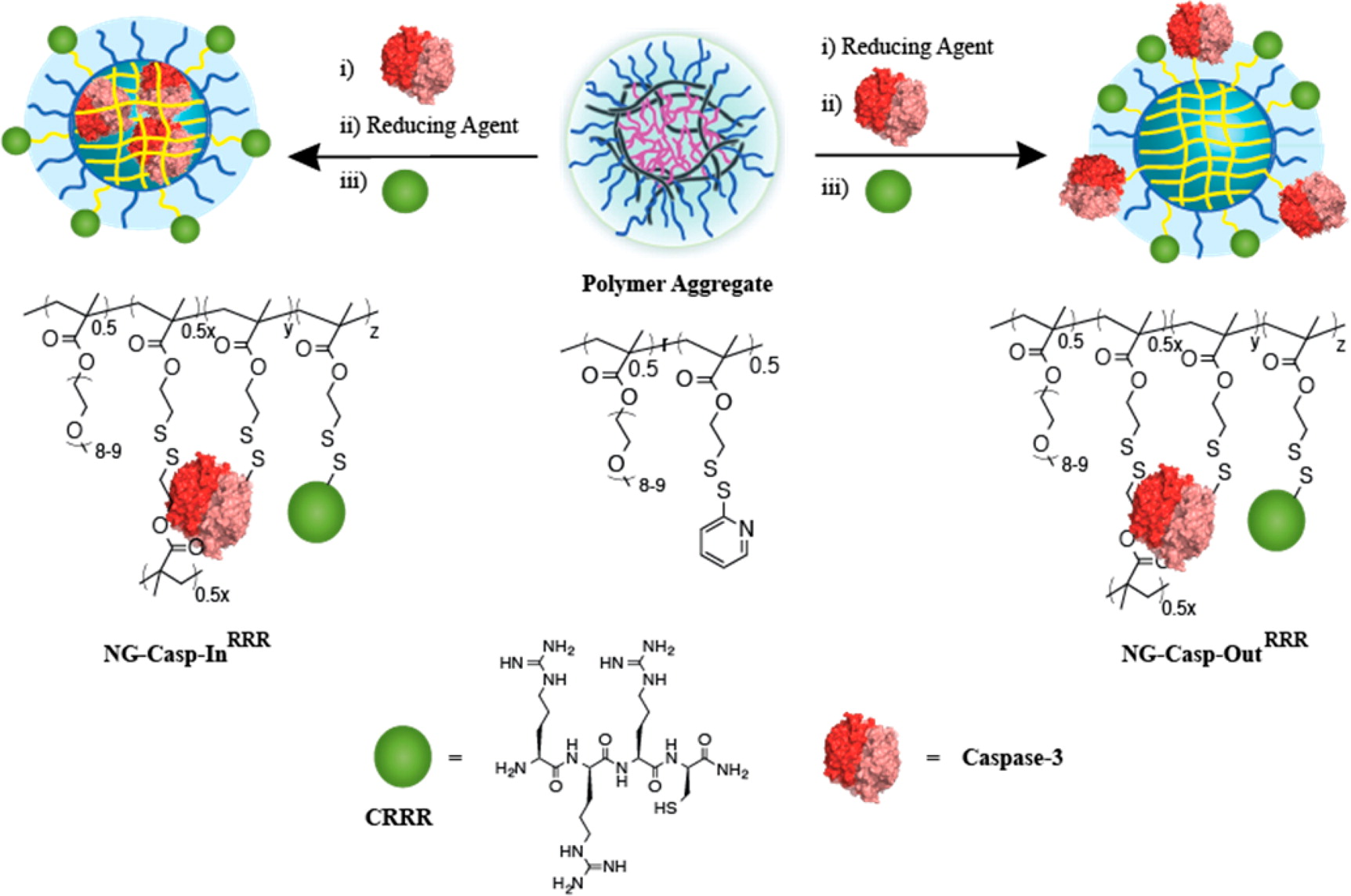
Caspase Delivery
Hijacking apoptotic pathways of unregulated cell proliferation is an attractive and powerful therapeutic strategy. In the Hardy lab, we are interested in understanding the mechanisms and pathways of the various caspases, the executioners of apoptosis. To utilize the potential therapeutic roles of caspases, we have collaborated with the Thayumanavan lab and have developed a nanogel platform for intracellular caspase delivery. As caspases are unable to penetrate the cell membrane on their own, targeted delivery of these caspase-nanogels provides a method for specific activation of apoptosis in a desired population. We are working towards probing the caspase-nanogel conjugate self-assembly process to elucidate the mechanisms of cellular uptake, release and potency.